There is great current interest in exploiting ribosome chemistry to generate sequence-defined chemical polymers that extend beyond L-⍺-amino acids to new backbone modifications and polymerization chemistries. Although the E. coli ribosome tolerates certain non-L-⍺-amino acids in vitro, few structural insights are available, and the boundary conditions for efficient bond formation are unknown.
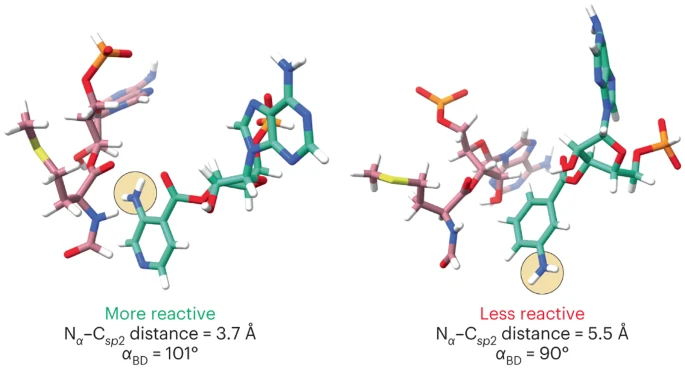
In this paper, the authors describe a 2.1 Å cryo-EM structure of the E. coli ribosome bound to well-resolved, acylated A- and P-site tRNAs and a metadynamics-based computational approach that evaluates how well non-L-⍺-amino acids are accommodated within the ribosome active site. The authors find that reactive monomers across diverse structural classes favor a conformational space characterized by an A-site nucleophile to P-site carbonyl distance of < 4 Å and a Bürgi-Dunitz angle of 90-110o. Monomers whose free energy minima fall outside these regions do not react. The computational workflow the authors describe should accelerate the design and the in vivo and in vitro ribosomal synthesis of sequence-defined, non-peptide heterooligomers.
This work was a collaboration between the labs of Scott Miller, Jamie Cate, and Alanna Schepartz and with Ara Abramyan of Schrödinger.
Read the paper: https://www.nature.com/articles/s41557-023-01226-w
Read the accompanying News and Views: https://www.nature.com/articles/s41557-023-01243-9
Read coverage of this paper in Berkeley News, Nature News